Using the BV-BRC Command-line Interface¶
BV-BRC is an integration of different types of data and software tools that support research on bacterial pathogens. The typical biologist seeking access to the BV-BRC data and tools will usually explore the web-based user interface. However, there are many instances in which programatic or command-line interfaces are more suitable. For users that wish command-line access to BV-BRC, we provide the tools described in this document. We call these tools the P3-scripts. They are intended to run on your machine, going over the network to access the services provided by BV-BRC.
Document Conventions¶
In this document, we observe the following conventions.
Text that you enter or type is shown in a white-background box.
This is input.
Output is shown in a yellow-background box. In general, you will only see the top portion of the output, since the whole thing could be quite large.
This is the top portion
of the output.
Output is usually tab-delimited, and you will see columns separated by multiple spaces that don’t always line up.
If it is necessary to show multiple excerpts of a single large output stream, the missing parts will be shown with a gray bar.
This is the top part.
This is somewhere in the middle.
NOTE: we add new genomes to the BV-BRC database every week. Your
results from the examples in this tutorial may not match ours.
Installing the CLI Release¶
Since the CLI tools run on your computer, to use them you will need to download and install a software package in order to use them.
We currently have macOS and Debian/Ubuntu releases of the BV-BRC Command Line Interface. A Windows version is in the works.
The releases are available at the BV-BRC github site. Full installation installations are available in Installing the BV-BRC Command Line Interface.
Command-Line Help¶
You can specify --help as a command-line option on any command to
get a summary of the options and parameters, for example
p3-match --help
p3-match.pl [-bchiv] [long options...] match-value
-i STR --input STR name of the input file (if not the standard
input)
-c STR --col STR column number (1-based) or name
-b INT --batchSize INT input batch size
--nohead file has no headers
-v --invert --reverse output non-matching records
--discards STR name of file to contain discarded records
-h --help display usage information
The BV-BRC Database¶
The main BV-BRC database is organized as a series of large, heavily-indexed relational tables. From the perspective of the CLI, there are five main tables representing objects of interest, connected by four relationships.
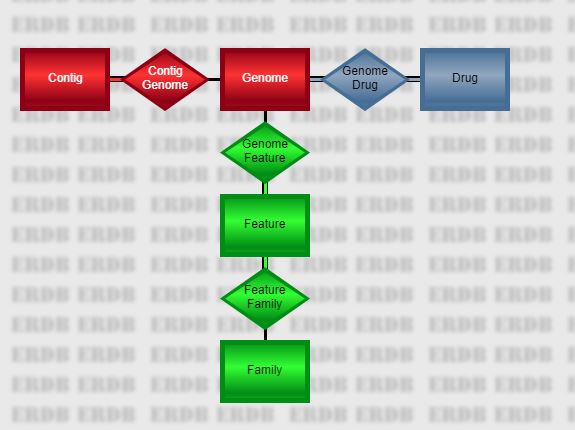
The five entities are as follows.
- Genome
A genome is a set of contigs and annotations representing our best estimate of the DNA sequence for an organism. Use p3-all-genomes to list all of the genomes or a subset. Given a list of genomes,
Use p3-get-genome-data to retrieve data about the individual genomes.
Use p3-get-genome-features to access the features of the genomes.
Use p3-get-genome-contigs to access the genomes’ sequences.
Use p3-get-genome-drugs to access drug resistance data about the genomes.
Fields from the Genome table appear in the output with a heading prefix of
genome. Thus, the genome_name will be in a column namedgenome.genome_name.- Contig
Represents one of the DNA sequences that comprise a genome. A contig can be a chromosome, a plasmid, or a fragment thereof. Contig data can be accessed from genome IDs using p3-get-genome-contigs. Fields from the Contig table appear in the output with a heading prefix of
contig. Thus, the length will be in a column namedcontig.length.- Drug
Represents an antimicrobial drug used for therapeutic treatment. This table is the anchor for all antimicrobial resistance data in BV-BRC. Use p3-all-drugs to get a list of drugs. Use p3-get-drug-genomes to get resistance data relating to specific drugs from a list. Fields from the Drug table appear in the output with a heading prefix of
drug. Thus, the molecular_formula will be in a column nameddrug.molecular_formula.- Feature
Represents a region of interest in a genome. This could be a CRISPR array, an RNA site, a protein encoding region, or a regulatory site, among others. A feature can be split across multiple regions, or even multiple contigs, but never multiple genomes. Given a list of genome IDs, use p3-get-genome-features to access the features in the genomes. Given a list of family IDs, use p3-get-family-features to access the features in the families. Given a list of feature IDs, use p3-get-feature-data to access data about those features. It is important to remember that the ID of the feature is called *patric_id*, not *feature_id*. The internal feature ID is a long string with a lot of data packed into it that may change if the genome is re-annotated (e.g.
PATRIC.269798.23.NC_008255.CDS.22581.24344.fwd). The patric_id value is shorter and more consistent (fig|269798.23.peg.22). Fields from the Feature table appear in the output with a heading prefix offeature. Thus, the location will be in a column namedfeature.location.- Family
Represents a protein family, which is a set of features believed to be isofunctional homologs. Given a list of family IDs, use p3-get-family-features to get data about the features in the families or p3-get-family-data to get data about the families themselves. Given a list of feature IDs, use p3-get-feature-data to get the families to which the features belong. There are three types of protein families supported– local families which are confined to one genus, global families which cross the entire database, and figfams, which are computed using a different method. Fields from the Family table appear in the output with a heading prefix of
family. Thus, the product will be in a column namedfamily.product.
Files and Pipelines¶
The BV-BRC CLI operates on tab-delimited files. That is, each record is divided into fields or columns separated by tab characters. The first record in each file contains the name of each column. Typically, a column name consists of a record name, a dot, and a field name. For example, the following file fragment contains a column from the genome table followed by two columns from the feature table.
genome.genome_id feature.patric_id feature.product
670.470 fig|670.470.repeat.1 repeat region
670.470 fig|670.470.repeat.2 repeat region
670.470 fig|670.470.repeat.3 repeat region
670.470 fig|670.470.rna.1 tRNA-Ala
670.470 fig|670.470.rna.2 tRNA-Ile
670.470 fig|670.470.repeat.4 repeat region
670.470 fig|670.470.rna.3 16S ribosomal RNA
670.470 fig|670.470.repeat.5 repeat region
670.470 fig|670.470.rna.4 tRNA-Val
670.470 fig|670.470.rna.5 tRNA-Ala
670.470 fig|670.470.repeat.6 repeat region
The scripts are designed so they can be chained together in pipelines where the output of one becomes input to the next. For example, the above file was generated by the pipeline
p3-all-genomes --eq genus,Methylobacillus | p3-get-genome-features --attr patric_id --attr product
In the first command of this pipeline, the --eq command-line option
was used to filter a query, while the --attr option in the second
command was used to specify the output columns and the order in which
they appear. These options are available on all of the database scripts.
For get-type scripts (p3-get-genome-data,
p3-get-feature-data, …), you must supply the id of the
object of interest, e.g., the genome id, feature id, etc. By default,
the last column in the input file is used as the key field for these
get-type scripts. You can modify this behavior using the --col
command-line option. The special value 0 denotes the last column,
but you can also use a 1-based column number (1 for the first, 2
for the second) or a column name. If the field-name portion of the
column name is unique, you can leave off the table-name portion. So, if
you want to get location information from the features output by the
pipeline above (identified in column feature.patric_id, which is the
second one), you could use any of the three following commands
p3-get-feature-data --col=feature.patric_id --attr sequence_id --attr location <input.tbl
p3-get-feature-data --col=2 --attr sequence_id --attr location <input.tbl
p3-get-feature-data --col=patric_id --attr sequence_id --attr location <input.tbl
where input.tbl is the above output file.
The special script p3-extract allows you to select columns from a file and even change the order. Thus, the following pipeline removes the genome ID from our file and puts the feature ID at the end before asking p3-get-feature-data for the location information.
p3-extract feature.product feature.patric_id <input.tbl | p3-get-feature-data --attr sequence_id --attr location
The same flexibility provided for arguments of the --col option is
available anywhere you specify column names, including the parameters of
p3-extract. So, the following invocation is equivalent to the above.
p3-extract product 2 <input.tbl | p3-get-feature-data --attr sequence_id --attr location
Because of the presence of the headings, many standard file-manipulation commands won’t work the way you expect. For example, if you use a standard sort command, the headers will sort somewhere into the middle of the file. We provide p3 scripts for several of the most common needs.
P3 Script |
Description |
Unix Equivalent |
|---|---|---|
p3-extract |
Select and re-order specific columns. |
cut |
p3-sort |
Sort by specified columns. |
sort |
p3-match |
Select records that possess (or do not possess) a particular value in a specified column. |
grep |
p3-join |
Horizontally join two files on a single key field. |
join |
p3-head |
Display the first few lines of a file. |
head |
p3-echo |
Create a small file. |
echo |
These commands do not work precisely like their unix equivalents. Most have fewer options: for example, p3-match searches for text in a single column rather than the entire file and does not support regular expressions.
p3-echo¶
The p3-echo command is your most important tool for creating small
files that feed into pipes. The --title command-line option
(abbreviated -t) allows you to specify the title for the column you
are creating. Each positional parameter forms a single record with a
single column.
p3-echo -t genome_id 1313.7001 1313.7002 1313.7016
genome_id
1313.7001
1313.7002
1313.7016
You can create a multi-column file by specifying multiple titles. There will be one output column for each title specified. In the example below, there are three titles, so the output table is three columns. Every triple of parameters produces a record.
p3-echo -t genome_id -t sequences -t gc_content 1313.7001 52 39.64 1313.7002 45 39.63 1313.7016 58 39.77
genome_id sequences gc_content
1313.7001 52 39.64
1313.7002 45 39.63
1313.7016 58 39.77
If a field contains special characters such as spaces or pipe symbols, use double quotes to ensure the characters are interpreted correctly.
p3-echo -t genome_id -t patric_id -t product 1313.7001 "fig|1313.7001.peg.1362" "hypothetical protein"
genome_id patric_id product
1313.7001 fig|1313.7001.peg.1362 hypothetical protein
If you leave off the title parameter, the default title id is used.
This is a handy shortcut when you’re in a hurry.
p3-echo 1313.7001 1313.7016
id
1313.7001
1313.7016
Of course, you can only do that for a single-column output.
Database Script Examples¶
In this section we briefly discuss the main database scripts.
- p3-all-genomes
This script lists all genomes with various characteristics. For example,
p3-all-genomes --eq genome_name,Streptomyces
genome.genome_id 284037.4 67257.17 68042.5 68042.6 1395572.3 68570.5 1160718.3 749414.3 66876.3 249567.6
would list all genomes in the genus Streptomyces. (That is, all genomes whose names start with that word.) The
--eqparameter introduces an equality constraint. In BV-BRC, string searches perform a word-based substring match, which allows us to easily do queries of this type. The various database commands all support the--eqoption. In addition, you can specify output fields using the--attroption. Thus,p3-all-genomes --eq genome_name,Streptomyces --attr genome_id --attr genome_name
would output both the ID and name of each genome found, as shown below.
genome.genome_id genome.genome_name 284037.4 Streptomyces sporocinereus strain OsiSh-2 67257.17 Streptomyces albus subsp. albus strain NRRL F-4371 68042.5 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 16556 68042.6 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 13472 1395572.3 Streptomyces albulus PD-1 68570.5 Streptomyces albulus strain NK660 1160718.3 Streptomyces auratus AGR0001 749414.3 Streptomyces bingchenggensis BCW-1 66876.3 Streptomyces chattanoogensis strain NRRL ISP-5002 249567.6 Streptomyces decoyicus strain NRRL 2666
To get a complete list of the available fields, use the
--fieldsoption. This option is available for all the database scripts described in this section.p3-all-genomes --fields
_version_ additional_metadata (multi) altitude antimicrobial_resistance (multi) antimicrobial_resistance_evidence assembly_accession assembly_method bioproject_accession biosample_accession biovar
The parenthetical
(multi)indicates that a field has multiple values. The values returned will be separated by the current delimiter (which defaults to a double colon::). There is also a parenthetical(derived)which indicates the value of the field is computed from other fields and can’t be used in filtering.- p3-get-genome-data
Given an input file of genome IDs, this script allows you to retrieve additional data and fields. For example, the following pipeline gets all Streptomyces genomes and then appends the genome name, number of contigs, and DNA length.
p3-all-genomes --eq genome_name,Streptomyces | p3-get-genome-data --attr genome_name --attr contigs --attr genome_length
genome.genome_id genome.genome_name genome.contigs genome.genome_length 284037.4 Streptomyces sporocinereus strain OsiSh-2 125 10242506 67257.17 Streptomyces albus subsp. albus strain NRRL F-4371 307 9246299 68042.5 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 16556 133 10141569 68042.6 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 13472 680 9464604 1395572.3 Streptomyces albulus PD-1 425 9340057 68570.5 Streptomyces albulus strain NK660 9372401 1160718.3 Streptomyces auratus AGR0001 213 7825489 749414.3 Streptomyces bingchenggensis BCW-1 0 11936683 66876.3 Streptomyces chattanoogensis strain NRRL ISP-5002 217 9129105
In actual fact, the use of p3-get-genome-data in the above pipeline is redundant, since p3-all-genomes supports the same command-line options. In practice, you will use p3-get-genome-data to process genome ID files created on a separate occasion or via other scripts that don’t have the full power of p3-all-genomes. If you don’t specify any
--attrvalues, you get the same output fields as found on the BV-BRC genome list tab.p3-all-genomes --eq genome_name,Streptomyces | p3-get-genome-data
genome.genome_id genome.genome_name genome.genome_id genome.genome_status genome.sequences genome.patric_cds genome.isolation_country genome.host_name genome.disease genome.collection_year genome.completion_date 284037.4 Streptomyces sporocinereus strain OsiSh-2 284037.4 WGS 125 9060 China Rice 2012 2016-08-16T00:00:00Z 67257.17 Streptomyces albus subsp. albus strain NRRL F-4371 67257.17 WGS 307 8633 2016-01-26T00:00:00Z 68042.5 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 16556 68042.5 WGS 133 8955 2016-02-05T00:00:00Z 68042.6 Streptomyces hygroscopicus subsp. hygroscopicus strain NBRC 13472 68042.6 WGS 680 8767 2016-02-05T00:00:00Z 1395572.3 Streptomyces albulus PD-1 1395572.3 WGS 482 8332 China 2013-12-05T00:00:00Z 68570.5 Streptomyces albulus strain NK660 68570.5 Complete 2 8793 China 2014-06-18T00:00:00Z 1160718.3 Streptomyces auratus AGR0001 1160718.3 WGS 238 6866 China 2012-07-23T00:00:00Z 749414.3 Streptomyces bingchenggensis BCW-1 749414.3 Complete 1 10313 China 2010-05-28T00:00:00Z 66876.3 Streptomyces chattanoogensis strain NRRL ISP-5002 66876.3 WGS 217 8838 United States 2015-09-18T00:00:00Z 249567.6 Streptomyces decoyicus strain NRRL 2666 249567.6 WGS 304 8231 United States 2015-08-19T00:00:00Z 1907.4 Streptomyces glaucescens GLA.O 1907.4 Complete 2 6719 India 2014-10-01T00:00:00Z 1172567.3 Streptomyces globisporus C-1027 1172567.3 WGS 278 6980 China 2012-05-04T00:00:00Z
This is typical of the
p3-getscripts: the default attributes match what you see on the web site as closely as possible.- p3-get-genome-features
Given a file of genome IDs, return data about the genome’s features. So, for example, the following pipeline would return the ID and function (product) of each feature in the Cytophaga genomes.
p3-all-genomes --eq genus,Cytophaga | p3-get-genome-features --attr patric_id --attr product
genome.genome_id feature.patric_id feature.product 269798.23 fig|269798.23.peg.1 hypothetical protein 269798.23 fig|269798.23.peg.2 Capsular polysaccharide synthesis enzyme Cap8C; Manganese-dependent protein-tyrosine phosphatase (EC 3.1.3.48) 269798.23 fig|269798.23.peg.3 Dihydroflavonol-4-reductase (EC 1.1.1.219) 269798.23 fig|269798.23.peg.4 TPR domain protein 269798.23 fig|269798.23.peg.5 Phosphosulfolactate synthase (EC 4.4.1.19) 269798.23 fig|269798.23.peg.6 DedA protein 269798.23 fig|269798.23.peg.7 Shikimate 5-dehydrogenase I alpha (EC 1.1.1.25) 269798.23 fig|269798.23.peg.8 hypothetical protein 269798.23 fig|269798.23.peg.9 Excinuclease ABC subunit B 269798.23 fig|269798.23.peg.10 DNA polymerase III epsilon subunit 269798.23 fig|269798.23.peg.11 hypothetical protein 269798.23 fig|269798.23.peg.12 putative fatty acid hydroxylase
You can use the
--fieldsoption to list all the fields available in a feature record. In addition, you have access to the usual filtering parameters–--eqas well as--lt,--gt,--le,--ge, and--ne. So, for example, the following command would restrict the features to CDS features of at least 500 base pairs.p3-all-genomes --eq genus,Cytophaga | p3-get-genome-features --eq feature_type,CDS --ge na_length,500 --attr patric_id --attr product
genome.genome_id feature.patric_id feature.product 269798.23 fig|269798.23.peg.2 Capsular polysaccharide synthesis enzyme Cap8C; Manganese-dependent protein-tyrosine phosphatase (EC 3.1.3.48) 269798.23 fig|269798.23.peg.3 Dihydroflavonol-4-reductase (EC 1.1.1.219) 269798.23 fig|269798.23.peg.4 TPR domain protein 269798.23 fig|269798.23.peg.5 Phosphosulfolactate synthase (EC 4.4.1.19) 269798.23 fig|269798.23.peg.6 DedA protein 269798.23 fig|269798.23.peg.7 Shikimate 5-dehydrogenase I alpha (EC 1.1.1.25) 269798.23 fig|269798.23.peg.8 hypothetical protein 269798.23 fig|269798.23.peg.9 Excinuclease ABC subunit B 269798.23 fig|269798.23.peg.10 DNA polymerase III epsilon subunit 269798.23 fig|269798.23.peg.12 putative fatty acid hydroxylase 269798.23 fig|269798.23.peg.13 Glycosyl transferase
- p3-get-genome-contigs
Given a file of genome IDs, returns the contigs. The following pipeline returns all the contigs in genome 28903.66.
p3-echo -t genome_id 28903.66 | p3-get-genome-contigs --attr sequence_id --attr sequence
The output will have three columns, including the genome ID, the ID of the contig, and the actual DNA sequence (which can be quite long). Again, use the
--fieldsoption to see which fields are available for output and filtering in the contig records.- p3-get-genome-drugs
Given a file of genome IDs, output the drug resistance information we have on those genomes. For many genomes, no such data is yet available, so it is not hard to get an empty file output from this command. The following pipeline outputs the default resistance data information for all the Acinetobacter pittii genomes.
p3-all-genomes --eq "genome_name,Acinetobacter pittii" | p3-get-genome-drugs
genome.genome_id genome_drug.genome_id genome_drug.antibiotic genome_drug.resistant_phenotype 48296.102 48296.102 meropenem Resistant 48296.102 48296.102 imipenem Resistant 48296.102 48296.102 ciprofloxacin Susceptible 48296.102 48296.102 gentamicin Susceptible 48296.102 48296.102 amikacin Susceptible 48296.102 48296.102 tigecycline 48296.104 48296.104 imipenem Resistant 48296.104 48296.104 ciprofloxacin Susceptible 48296.104 48296.104 gentamicin Susceptible
- p3-all-drugs
This script lists anti-microbial drugs from the database. Use the
--fieldsoption to see a list of all the fields you can select. The default is to simply list the antibiotic name, as shown below.p3-all-drugs
drug.antibiotic_name amikacin amoxicillin amoxicillin/clavulanic acid ampicillin ampicillin/sulbactam azithromycin aztreonam bacitracin capreomycin cefaclor cefazolin
- p3-get-drug-genomes
Given a file of antibiotic names, display all the resistance data for those antibiotics. The following pipeline lists all the genomes resistant to at least one drug.
p3-all-drugs | p3-get-drug-genomes --attr genome_id --attr genome_name --resistant
drug.antibiotic_name genome_drug.genome_id genome_drug.genome_name amikacin 1304920.3 Klebsiella pneumoniae 361_1301 amikacin 1427177.3 Mycobacterium tuberculosis XTB13-081 amikacin 1427178.3 Mycobacterium tuberculosis XTB13-082 amikacin 1427180.3 Mycobacterium tuberculosis XTB13-084 amikacin 1427185.3 Mycobacterium tuberculosis XTB13-091 amikacin 1427191.3 Mycobacterium tuberculosis XTB13-097 amikacin 1427192.3 Mycobacterium tuberculosis XTB13-098 amikacin 1427193.3 Mycobacterium tuberculosis XTB13-100 amikacin 1427199.3 Mycobacterium tuberculosis XTB13-107 amikacin 1427200.3 Mycobacterium tuberculosis XTB13-108 amikacin 1427202.3 Mycobacterium tuberculosis XTB13-110 amikacin 1427204.3 Mycobacterium tuberculosis XTB13-112 amikacin 1427207.3 Mycobacterium tuberculosis XTB13-115
Rather than typing
--eq resistant_phenotype,resistant, the p3-get-drug-genomes script provides the special command-line options--resistantand--susceptibleto filter for the appropriate resistance phenotypes automatically.- p3-get-family-features
Given a list of protein family IDs, get all the features in the families. BV-BRC supports three types of protein families–
local,global, andfigfam. The--ftypeparameter specifies the type of family desired. So, for example, the following pipeline finds the global family for the feature fig|1105121.3.peg.460 and then lists the ID and product of each family member.p3-echo -t feature_id "fig|1105121.3.peg.460" | p3-get-feature-data --attr pgfam_id | p3-get-family-features --ftype=global --attr patric_id --attr product
Note that the features found are listed in the column feature.patric_id, while the original feature is maintained in the first column feature_id.
feature_id feature.pgfam_id feature.patric_id feature.product fig|1105121.3.peg.460 PGF_00112374 fig|1313.8637.peg.2087 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.8636.peg.1563 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.8645.peg.110 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.12423.peg.2037 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1330044.3.peg.533 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5699.peg.1778 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5750.peg.307 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5754.peg.739 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5758.peg.1823 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5781.peg.1819 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5778.peg.686 hypothetical protein fig|1105121.3.peg.460 PGF_00112374 fig|1313.5729.peg.1554 hypothetical protein
- p3-get-feature-data
Given a file of feature IDs, return data from those features. Again, use the
--fieldsoption to list the fields you can use for filtering and display. The following pipeline lists the function (product) and protein sequence of each peg of less than 300 base pairs in the genome 1105121.3.p3-echo -t genome_id 1105121.3 | p3-get-genome-features --lt na_length,300 --eq feature_type,CDS --attr patric_id | p3-get-feature-data --attr product --attr aa_sequence
genome_id feature.patric_id feature.product feature.aa_sequence 1105121.3 fig|1105121.3.peg.1487 BOX elements MKIKEQTRKLAASCSKHCFEVVDKTDEVSYIYNPRRR 1105121.3 fig|1105121.3.peg.1508 hypothetical protein MISTTYRNHRKRFGLRMNLIAEKVSKTLDKTFDKDVREIPTSQFYQKFVDEMGRTYSGNLILQELITVNGAYKATYIGELSSN 1105121.3 fig|1105121.3.peg.1557 hypothetical protein MKREVISNGNDGPSQEILIFTKQIRHWILSDQVISGKRKLFFREDTPKEILDLYENIKSKLDFAYQEVYSNNGLKKYEK 1105121.3 fig|1105121.3.peg.1598 BOX elements MKIKEQTRKLAAGCSKHCFEVVDRTDEVSNLHTARRR 1105121.3 fig|1105121.3.peg.1776 hypothetical protein MVASASASSTSTQAQEQVDKSELRALSQELDQRLKALATVSDPKIDATKAVLLDAQKAPEDSALTE 1105121.3 fig|1105121.3.peg.10 hypothetical protein MENLLDVIEQFLGLSDEKLEELADKNQLLRLQEEKERKNA 1105121.3 fig|1105121.3.peg.94 BOX elements MKIKEQTRKLAAGCSKHCFEVVDKTDEVSYIYLRQGEADAV 1105121.3 fig|1105121.3.peg.220 Ribonucleotide reductase of class III (anaerobic), large subunit (EC 1.17.4.2) MVKRTCGYLGNPQARPMVNGRHKEIAARVKHMNGSTIKIAGHQVTN 1105121.3 fig|1105121.3.peg.228 LSU ribosomal protein L23p (L23Ae) MNLYDVIKKPVITESSMAQLEAGKYVFEVDTRAHKLLIKQAVEAAFEGVKVANVNTINVKPKAKRVGRYTGFTNKTKKAIITLTADSKAIELFAAEAE 1105121.3 fig|1105121.3.peg.230 SSU ribosomal protein S19p (S15e) MGRSLKKGPFVDEHLMKKVEAQANDEKKKVIKTWSRRSTIFPSFIGYTIAVYDGRKHVPVYIQEDMVGHKLGEFAPTRTYKGHAADDKKTRRK
The use of p3-get-feature-data here is redundant, since you could get the same result by placing the attribute requests directly on p3-get-genome-features.
p3-echo -t genome_id 1105121.3 | p3-get-genome-features --lt na_length,300 --eq feature_type,CDS --attr patric_id --attr product --attr aa_sequence
p3-get-feature-data is provided for the situation where you are piping in the feature list from something external or precomputed.
What Is a BV-BRC Workspace?¶
Users of BV-BRC have access to a wealth of public data that support interpretation of prokaryotic genomes. The BV-BRC team actively integrates newly-sequenced genomes, data relating to antimicrobial resistance, expression data, pathway data and subsystem data into an integrated framework that can be queried using either the BV-BRC UI or the CLI.
In the BV-BRC UI, your workspace looks a lot like a standard file system, divided into folders full of data. In addition to files you upload, such as FASTA and FASTQ files, there will also be typed objects such as genomes, feature groups, and genome groups. The CLI allows you to move these typed objects between your workspace and your file system so you can manipulate them at will.
Logging In¶
To access your workspace, you need a BV-BRC account. If you do not have one already, go to https://user.bv-brc.org/register and register now.
Now that you have a working user name and password, you can use the
p3-login script to tell the CLI who you are. For example, if your
name is rastuser25, you would type
p3-login rastuser25
The script asks you for your password and places a special file on your hard drive that can be used to get authorized access to your workspace data. To log out again, simply use
p3-login --logout
At any time, you can verify your login status using
p3-login --status
If you are logged out, it will respond
You are currently logged out of BV-BRC.
If you are logged in, you will get something like
You are logged in as rastuser25@patricbrc.org.
Working with Genome Groups¶
Your workspace looks like a full-blown file system, but there are three special folders.
Genome Groups contains named lists of genomes.
Feature Groups contains named lists of features.
QuickData contains folders full of genomes you submitted through the CLI annotation interface.
To create a genome group, you use p3-put-genome-group. Say, for example, you want to examine Streptococcus penumoniae genomes that are resistant to penicillin. The following query command will return this list of genomes (we will discuss all query commands in more details later).
p3-echo -t antibiotic penicillin | p3-get-drug-genomes --eq "genome_name,Streptococcus pneumoniae" --resistant --attr genome_id --attr genome_name >resist.tbl
This particular command asks for data from the anti-microbial resistance
table. Each record in this table posits a relationship between a genome
and an antibiotic drug. We are accessing the table from the direction of
taking a drug and finding resistant genomes. To do this, we need a file
with a drug name in it. The p3-echo command creates this file: the
-t antibiotic parameter tells it we want a one-column file with a
column header of antibiotic. We put the single record penicillin
in that column.
The antibiotic file is then piped into p3-get-drug-genomes. Its parameters do the following.
--eq "genome_name,Streptococcus pneumoniae"Only include records for Streptococcus pneumoniae genomes. Because this is a string field, it does a substring match. A genome name including follow-on strain information (e.g.
Streptococcus pneumoniae strain LMG2888) will still match.--resistantOnly include records that state the genome is resistant. This is a special parameter for the p3-get-drug-genomes and p3-get-genome-drugs commands that is provided for convenience.
--attr genome_idOutput the genome ID.
--attr genome_nameOutput the genome name.
When the command completes, the file resist.tbl will contain around 114 lines beginning with the following.
antibiotic genome_drug.genome_id genome_drug.genome_name
penicillin 1313.7006 Streptococcus pneumoniae P310010-154
penicillin 1313.7016 Streptococcus pneumoniae P310937-212
penicillin 1313.7018 Streptococcus pneumoniae P311313-217
penicillin 760749.3 Streptococcus pneumoniae GA05248
penicillin 760763.3 Streptococcus pneumoniae GA11304
penicillin 760765.3 Streptococcus pneumoniae GA11663
penicillin 760766.3 Streptococcus pneumoniae GA11856
penicillin 760769.3 Streptococcus pneumoniae GA13338
penicillin 760771.3 Streptococcus pneumoniae GA13455
penicillin 760776.3 Streptococcus pneumoniae GA14373
penicillin 760777.3 Streptococcus pneumoniae GA14688
Now we want to create a group for these genomes called resist_strep. We use the following command.
p3-put-genome-group --col=2 resist_strep <resist.tbl
The --col=2 tells the command that the genome IDs are in the second
column. The genome group is simply a set of genome IDs, so the other
columns will be ignored by the command. You can read the group back at
any time using p3-get-genome-group.
p3-get-genome-group resist_strep
Will output
resist_strep.genome_id
1313.7006
1313.7016
1313.7018
760749.3
760763.3
760765.3
760766.3
760769.3
760771.3
and so on. Note that if you want to see the names as well, you can use the p3-get-genome-data command to add them
p3-get-genome-group resist_strep | p3-get-genome-data --attr genome_name
resist_strep.genome_id genome.genome_name
1313.7006 Streptococcus pneumoniae P310010-154
1313.7016 Streptococcus pneumoniae P310937-212
1313.7018 Streptococcus pneumoniae P311313-217
760749.3 Streptococcus pneumoniae GA05248
760763.3 Streptococcus pneumoniae GA11304
760765.3 Streptococcus pneumoniae GA11663
760766.3 Streptococcus pneumoniae GA11856
760769.3 Streptococcus pneumoniae GA13338
760771.3 Streptococcus pneumoniae GA13455
760776.3 Streptococcus pneumoniae GA14373
760777.3 Streptococcus pneumoniae GA14688
Next we will ask for the genomes that are susceptible to penicillin. We
use the same command as before except we put susceptible in place of
resistant. We’re going to pipe the results directly into
p3-put-genome-group to store them in our workspace.
p3-echo -t antibiotic penicillin | p3-get-drug-genomes --eq "genome_name,Streptococcus pneumoniae" --susceptible --attr genome_id --attr genome_name | p3-put-genome-group --col=2 weak_strep
Now when you ask for the group back, you would get something like the following.
p3-get-genome-group weak_strep
weak_strep.genome_id
1313.6939
1313.6941
1313.6942
1313.6944
1313.6947
1313.7001
1313.7002
1313.7007
1313.7009
Working with Feature Groups¶
We want to look at features in the resistant Streptococcus pneumoniae genomes that distinguish them from the susceptible ones. Then we will gather those features into a feature group and store them in our workspace so we can work with them later. We have the genomes we want stored in the genome groups weak_strep and resist_strep. The command that processes them is called p3-signature-families.
p3-signature-families compares two genome groups– group 1 contains genomes that are interesting for some reason, group 2 contains genomes that are not. We can pipe one of the two groups directly into the command, but the other needs to be in a file. We will start by creating a file called weak.tbl that contains the weak_strep genomes.
p3-get-genome-group weak_strep >weak.tbl
Now we pipe in resist_strep (the interesting set) and specify weak.tbl as the source of group 2.
p3-get-genome-group resist_strep | p3-signature-families --gs2=weak.tbl >families.tbl
The output contains protein families that are common in the interesting set (resistant to penicillin) but not in the other set. If a set file is not specified, it is taken from the standard input. In this case, that would be the interesting set, since there is no –gs1 parameter.
Our signature families analysis script has no output, because we
redirected it to families.tbl. We can peek at the results using the
--all option of p3-extract.
p3-extract --all <families.tbl
counts_in_set1 counts_in_set2 family.family_id family.product
92 10 PGF_00112374 hypothetical protein
92 10 PGF_00303700 hypothetical protein
92 10 PGF_03497231 hypothetical protein
91 10 PGF_03497236 hypothetical protein
We found four protein families. The next step is to convert the families into feature IDs. The p3-get-family-features script performs that function. We will use the following command.
p3-get-family-features --gFile=resist.tbl --gCol=2 --ftype=global --col=family.family_id <families.tbl
The parameters work as follows.
- –gFile=resist.tbl
Only features from the genomes listed in the file resist.tbl should be included in the output.
- –gCol=2
The genome IDs in resist.tbl are in the second column.
- --ftype=global
The family IDs in the input file are global families. (Local families and FIGfams are also supported, but the p3-signature-families script uses global families.)
- –col=family.family_id
The protein family IDs in the input file are in a column named
family.family_id.
The output looks something like this.
counts_in_set1 counts_in_set2 family.family_id family.product feature.patric_id feature.refseq_locus_tag feature.gene_id feature.plfam_id feature.pgfam_id feature.product
92 10 PGF_00112374 hypothetical protein fig|1105121.3.peg.460 SPAR163_0451 0 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1069626.3.peg.432 SPAR154_0430 0 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.6771.peg.1961 ERS013947_01920 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5503.peg.1279 ERS013945_01218 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5634.peg.1224 ERS013952_01170 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1069624.3.peg.437 SPAR151_0439 0 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1069628.3.peg.451 SPAR156_0440 0 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5669.peg.1163 ERS013960_01123 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.6725.peg.2115 ERS013931_02059 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5465.peg.1094 ERS013930_01056 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5418.peg.2058 ERS013923_02003 PLF_1301_00002060 PGF_00112374 hypothetical protein
92 10 PGF_00112374 hypothetical protein fig|1313.5645.peg.1124 ERS013964_01084 PLF_1301_00002060 PGF_00112374 hypothetical protein
We didn’t tell p3-get-family-features what attributes of the features to display, so it defaulted to the columns normally found on the BV-BRC web page Features tab. We don’t have time to examine these features in detail now, but we can put them in a feature group by piping them into p3-put-feature-group as follows.
p3-get-family-features --gFile=resist.tbl --gCol=2 --ftype=global --col=family.family_id <families.tbl | p3-put-feature-group --col=feature.patric_id resist_fids
In the p3-put-feature-group command, the --col=feature.patric_id
parameter tells the command that the feature IDs are in the column with
that heading, and resist_fids is the group name. When you decide to
examine the features in greater detail, you can pull back the feature
IDs using p3-get-feature-group.
p3-get-feature-group resist_fids
The output will look something like this.
resist_fids.patric_id
fig|1105121.3.peg.460
fig|1069626.3.peg.432
fig|1313.6771.peg.1961
fig|1313.5503.peg.1279
fig|1313.5634.peg.1224
fig|1069624.3.peg.437
fig|1069628.3.peg.451
fig|1313.5669.peg.1163
fig|1313.6725.peg.2115
At any time, you can get a complete list of the groups in your workspace using the p3-list-genome-groups command or the p3-list-feature-groups command. So, if you have been following along the above examples and your workspace was empty before you began, you would see the following.
p3-list-genome-groups
resist_strep
weak_strep
p3-list-feature-groups
resist_fids
Extracting and Mining Genome Typed Objects (GTOs)¶
Sometimes you want to store a genome on your local hard drive. BV-BRC provides a special format for encapsulating all the data from a genome called the genome typed object or GTO. The p3-gto script allows you to download one or more BV-BRC genomes in GTO format. The following command downloads two strep genomes – 1313.7001 and 1313.7016– in GTO format and stores them in the current directory.
p3-gto 1313.7001 1313.7016
The GTO files have the same name as the genome ID with a suffix of
.gto. So, the above command creates 1313.7001.gto and
1313.7016 in the current directory. If you execute this command and
look at 1313.7001.gto, you will see something like the following
(with large portions in the middle removed).
{
"analysis_events" : [],
"scientific_name" : "Streptococcus pneumoniae P210774-233",
"source" : "PATRIC",
"source_id" : "1313.7001",
"id" : "1313.7001",
"taxonomy" : [
"cellular organisms",
"Bacteria",
"Terrabacteria group",
"Firmicutes",
"Bacilli",
"Lactobacillales",
"Streptococcaceae",
"Streptococcus",
"Streptococcus pneumoniae"
],
"contigs" : [
{
"genetic_code" : "11",
"dna" : "gaaaggacaaaatttgtcctttctcaagcttagctgacttcaacccactacagttgacaaagagcctgttttctcaataggattgtactcaggtgagtagggaggaagaggtaaaagtttatgcccaaactcttcacacaagagttctagcttacccattctatggaatcttgcattatccataataataaccgatggtgtggttaatgttggtaagagaaatttctgaaaccatacttcaaaaaagtcgctcgtcatcgtctcttcgtaagtcattggagcgattaattcaccatttgttagacctgcaaccaaagaaatcctctgatatcttcttccagatactttgcctcttcttaactgaccttttaatgagcgaccatattctcgataaaaataagtatcgaatcctgtttcgtcaatctaaacaggtgctaggtgctttaaactattaaaattcttaagaaataaggctacttatcgccctgaatatcaaaaaagaaaggacaaaatttgtcctttctcaagcttagctgacttcaacccactacagttgacaaagagcctgttttctcaataggattgtactcaggtgagtagggaggaagaggtaaaagtttatgcccaaactcttcacacaagagttctagcttacccattctatggaatcttgcattatccataataataaccgatggtgtggttaatgttggtaagagaaatttctgaaaccatacttcaaaaaagtcgctcgtcatcgtctcttcgtaagtcattggagcgattaattcaccatttgttagacctgcaaccaaagaaatcctctgatatcttcttccagatactttgcctcttcttaactgaccttttaatgagcgaccatattctcgataaaaataagtatcgaatcctgtttcgtcaatctaaacaggtgctaggtgctttaaactattaaaattcttaagaaataaggctactttttctgggtcttgttcatagtaggtgtggttctttttttcgagtgtagcccatagctttgagcgcatagtggatggtagttggatgacagccaaattcagaagctatttcagtcaaataagcgtct",
"id" : "1313.7001.con.0001"
},
{
"id" : "1313.7001.con.0002",
"genetic_code" : "11",
"dna" : "aaagaagctgttcgaaaagtaggcgatggttatgtctttgaggagaatggagtttctcgttatatcccagccaaggatctttcagcagaaacagcagcaggcattgatagcaaactggccaagcaggaaagtttatctcataagctaggagctaagaaaactgacctcccatctagtgatcgagaattttacaataaggcttatgacttactagcaagaattcaccaagatttacttgataataaaggtcgacaagttgattttgaggctttggataacctgttggaacgactcaaggatgtctcaagtgataaagtcaagttagtggatgatattcttgccttcttagctccgattcgtcatccagaacgtttaggaaaaccaaattcgcaaattacctacactgatgatgagattcaagtagccaagttggcaggcaagtacacaacagaagacggttatatctttgatcctcgtgatataaccagtgatgagggggatgcctatgtaactccacatatgacccatagccactggattaaaaaagatagtttgtctgaagctgagagagcggcagcccaggcttatgctaaagagaaaggtttgacccctccttcgacagaccatcaggatgcaggaaatactgaggcaaaaggagcagaagctatctacaaccgcgtgaaagcagctaagaaggtgccacttgatcgtatgccttacaatcttcaatatactgtagaagtcaaaaacggtagtttaatcatacctcattatgaccattaccataacatcaaatttgagtggtttgacgaaggcctttatgaggcacctaaggggtatactcttgaggatcttttggcgactgtcaagtactatgtcgaacatccaaacgaacgtccgcattcagataatggttttggtaacgctagcgaccatgttcaaagaaacaaaaatggtcaagctgataccaatcaaacggagaaacctcagacagaaaaacctgaggaagataaggaacatgatgaagtaagtgagccaactca"
},
],
"ncbi_taxonomy_id" : "1313",
"close_genomes" : [],
"domain" : "Bacteria",
"genetic_code" : "11",
"features" : [
{
"type" : "repeat_region",
"family_assignments" : [],
"annotations" : [
[
"Add feature from PATRIC",
"PATRIC",
1500218027.10933,
""
],
[
"Set function to repeat region",
"PATRIC",
1500218027.10933,
""
]
],
"aliases" : [],
"id" : "fig|1313.7001.repeat.1",
"function" : "repeat region",
"location" : [
[
"1313.7001.con.0001",
"67",
"+",
413
]
]
},
{
"location" : [
[
"1313.7001.con.0001",
"567",
"+",
539
]
],
"function" : "repeat region",
"id" : "fig|1313.7001.repeat.2",
"aliases" : [],
"annotations" : [
[
"Add feature from PATRIC",
"PATRIC",
1500218027.10944,
""
],
[
"Set function to repeat region",
"PATRIC",
1500218027.10944,
""
]
],
"family_assignments" : [],
"type" : "repeat_region"
},
]
}
This is a JSON-format string, which is to say, it displays an object with fields, some of which are other objects (denoted by curly braces) or lists (denoted by square brackets). JSON is a standard portable data format, described in detail here and supported by most programming languages. Without even fully understanding the notation, you can still see in the above listing that various bits of key metadata (scientific name, taxonomy, ID) are present in the file, along with the ID and sequence of each contig and various pertinent data about each feature.
You can use a minus sign (-) in the parameter list to specify that
the genome list come from the standard input. The following creates GTOs
for every genome in the genome group weak_strep.
p3-get-genome-group weak_strep | p3-gto -
This capability can be mixed with explicit genome IDs. So the following script creates a GTO for 594.8, all of the genomes in group weak_strep, and then genome 149539.441.
p3-get-genome-group weak_strep | p3-gto 594.8 - 149539.441
You can also use the --outDir option to specify that the output be
put in a different directory. The following creates a new subdirectory
PathogenGTO in the current directory and puts all the GTOs in it.
p3-get-genome-group weak_strep | p3-gto --outDir=PathogenGTO 594.8 - 149539.441
You are not required to write code to manipulate GTOs. Instead, we’ve included some useful scripts in the BV-BRC CLI. First and foremost is p3-gto-scan. For example, if you run
p3-gto-scan 1313.7001.gto
you would see the following analysis
Processing contigs of 1313.7001.gto.
Processing features of 1313.7001.gto.
All done.
contigs 52
dna 2101113
features 3382
functionAnalyzed 1418
functionRead 3382
functionReused 1964
roleMatch 1492
roleProcessed 3478
This rather arcane output tells you several things. First, that there are 52 contigs and 2,101,113 base pairs in the genome. It has 3382 features containing 1418 distinct assigned functions (functionAnalyzed). 3382 features had assigned functions (functionRead). This means every feature had a valid functional assignment, which is usually the case. 1964 of the features had redundant functions, that is, functions also found earlier in the genome (functionReused). 3478 roles were found (roleProcessed) of which 1492 were distinct (roleMatch).
If you want to see the actual roles, specify the command-line option
--verbose.
p3-gto-scan --verbose 1313.7001.gto
Role name 1313.7001.gto
(2E,6E)-farnesyl diphosphate synthase (EC 2.5.1.10) 1
1,2-diacylglycerol 3-glucosyltransferase (EC 2.4.1.337) 1
1,4-alpha-glucan (glycogen) branching enzyme, GH-13-type (EC 2.4.1.18) 1
1-phosphofructokinase (EC 2.7.1.56) 1
16S rRNA (cytidine(1402)-2'-O)-methyltransferase (EC 2.1.1.198) 1
16S rRNA (cytosine(1402)-N(4))-methyltransferase EC 2.1.1.199) 1
16S rRNA (cytosine(967)-C(5))-methyltransferase (EC 2.1.1.176) 1
16S rRNA (guanine(1207)-N(2))-methyltransferase (EC 2.1.1.172) 1
16S rRNA (guanine(527)-N(7))-methyltransferase (EC 2.1.1.170) 1
16S rRNA (guanine(966)-N(2))-methyltransferase (EC 2.1.1.171) 1
16S rRNA (uracil(1498)-N(3))-methyltransferase (EC 2.1.1.193) 1
In this table, each role name is shown along with the number of times it
occurs in the genome. You can see the features as well by adding the
--features command line.
p3-gto-scan --verbose --features 1313.7001.gto
Role name 1313.7001.gto Features containing role
(2E,6E)-farnesyl diphosphate synthase (EC 2.5.1.10) 1 fig|1313.7001.peg.1606
1,2-diacylglycerol 3-glucosyltransferase (EC 2.4.1.337) 1 fig|1313.7001.peg.679
1,4-alpha-glucan (glycogen) branching enzyme, GH-13-type (EC 2.4.1.18) 1 fig|1313.7001.peg.595
1-phosphofructokinase (EC 2.7.1.56) 1 fig|1313.7001.peg.227
16S rRNA (cytidine(1402)-2'-O)-methyltransferase (EC 2.1.1.198) 1 fig|1313.7001.peg.1813
16S rRNA (cytosine(1402)-N(4))-methyltransferase EC 2.1.1.199) 1 fig|1313.7001.peg.503
16S rRNA (cytosine(967)-C(5))-methyltransferase (EC 2.1.1.176) 1 fig|1313.7001.peg.1301
16S rRNA (guanine(1207)-N(2))-methyltransferase (EC 2.1.1.172) 1 fig|1313.7001.peg.83
16S rRNA (guanine(527)-N(7))-methyltransferase (EC 2.1.1.170) 1 fig|1313.7001.peg.1682
16S rRNA (guanine(966)-N(2))-methyltransferase (EC 2.1.1.171) 1 fig|1313.7001.peg.145
16S rRNA (uracil(1498)-N(3))-methyltransferase (EC 2.1.1.193) 1 fig|1313.7001.peg.728
Later on in this file you can see an example of a role that occurs in
multiple features. You will note that a double colon (::) is used to
separate the individual feature IDs.
6-phospho-beta-galactosidase (EC 3.2.1.85) 1 fig|1313.7001.peg.607
6-phospho-beta-glucosidase (EC 3.2.1.86) 4 fig|1313.7001.peg.1031::fig|1313.7001.peg.1517::fig|1313.7001.peg.443::fig|1313.7001.peg.896
6-phosphofructokinase (EC 2.7.1.11) 1 fig|1313.7001.peg.1372
6-phosphogluconate dehydrogenase, decarboxylating (EC 1.1.1.44) 1 fig|1313.7001.peg.542
This is a common convention in the BV-BRC CLI– when a single column
contains multiple values, we use a double colon to separate them. You
can use the --delim option to change this default. Supported
alternate delimiters include space, tab, and comma. For
example, the following would show if you coded --delim=space.
6-phospho-beta-galactosidase (EC 3.2.1.85) 1 fig|1313.7001.peg.607
6-phospho-beta-glucosidase (EC 3.2.1.86) 4 fig|1313.7001.peg.1031 fig|1313.7001.peg.1517 fig|1313.7001.peg.443 fig|1313.7001.peg.896
6-phosphofructokinase (EC 2.7.1.11) 1 fig|1313.7001.peg.1372
6-phosphogluconate dehydrogenase, decarboxylating (EC 1.1.1.44) 1 fig|1313.7001.peg.542
The true power in p3-gto-scan comes when you use it to compare multiple GTO files. The following command displays a summary of the differences between 1313.7001.gto and 1313.7016.gto.
p3-gto-scan 1313.7001.gto 1313.7016.gto
Processing contigs of 1313.7001.gto.
Processing features of 1313.7001.gto.
Processing contigs of 1313.7016.gto.
Processing features of 1313.7016.gto.
Role name 1313.7001.gto 1313.7016.gto
2,3-butanediol dehydrogenase, R-alcohol forming, (R)- and (S)-acetoin-specific (EC 1.1.1.4) 0 1
2-isopropylmalate synthase (EC 2.3.3.13) 1 2
23S rRNA (adenine(2058)-N(6))-dimethyltransferase (EC 2.1.1.184) => Erm(B) 0 1
4-hydroxybenzoate polyprenyltransferase and related prenyltransferases 0 1
5S rRNA 2 3
6-phospho-beta-galactosidase (EC 3.2.1.85) 1 2
AAA superfamily ATPase 0 1
ABC transporter amino acid-binding protein 0 1
ABC transporter, ATP-binding protein 13 11
ABC transporter, ATP-binding protein (cluster 3, basic aa/glutamine/opines) 3 4
ABC transporter, permease protein (cluster 3, basic aa/glutamine/opines) 5 6
ABC transporter, substrate-binding protein PebA (cluster 3, basic aa/glutamine/opines) 2 1
weak similarity to aminoglycoside phosphotransferase 1 0
* Features 3382 3304
* DNA 2101113 2052306
All done.
contigs 110
dna 4153419
features 6686
functionAnalyzed 1457
functionRead 6686
functionReused 5229
roleMatch 1356
roleMismatch 175
roleProcessed 6877
Only roles that differ between the two genomes are shown (175, the number in roleMismatch). For each, the role name is shown followed by the number of occurrences in 1313.7001.gto and then the number of occurrences in 1313.7016.gto. So, we can see that 2-isopropylmalate synthase occurs once in 1313.7001 but twice in 1313.7016. At the end of the role listing, feature and DNA counts are shown. We see that 1313.7016 has 78 fewer features and around 50,000 fewer base pairs (48,807 to be exact). 1356 roles occurred the same number of times in both genomes (roleMatch).
You can specify as many GTO file names as you wish in the parameter list
for p3-gto-scan. As with the single-genome case, --features
causes the features to be listed in the last column. The --verbose
option causes even the matching roles to be listed, so you can get
counts for everything.
The status and statistical messages are sent to the standard error output, and the role table to the standard output. Thus, if you redirect these to separate files, the direct output from p3-gto-scan can be used to get a convenient list of roles from the script. The file thus created is tab-delimited with headers, just like a normal CLI output file.
The script p3-gto-fasta creates FASTA files from a single GTO. Three command-line options (all mutually exclusive) are supported.
- --contig
Output a DNA fasta for the genome’s contigs. This is the default.
- --protein
Output a protein fasta for the genome’s features. Obviously, only protein-encoding features will be included.
- --feature
Output a DNA fasta for the genome’s features. All features are included.
You specify the name of the GTO file as the first parameter of p3-gto-fasta.
p3-gto-fasta 1313.7001.gto >1313.7001.fna
After this script, 1313.7001.fna will look something like this.
>1313.7001.con.0001
gaaaggacaaaatttgtcctttctcaagcttagctgacttcaacccactacagttgacaa
agagcctgttttctcaataggattgtactcaggtgagtagggaggaagaggtaaaagttt
atgcccaaactcttcacacaagagttctagcttacccattctatggaatcttgcattatc
cataataataaccgatggtgtggttaatgttggtaagagaaatttctgaaaccatacttc
aaaaaagtcgctcgtcatcgtctcttcgtaagtcattggagcgattaattcaccatttgt
tagacctgcaaccaaagaaatcctctgatatcttcttccagatactttgcctcttcttaa
ctgaccttttaatgagcgaccatattctcgataaaaataagtatcgaatcctgtttcgtc
aatctaaacaggtgctaggtgctttaaactattaaaattcttaagaaataaggctactta
tcgccctgaatatcaaaaaagaaaggacaaaatttgtcctttctcaagcttagctgactt
caacccactacagttgacaaagagcctgttttctcaataggattgtactcaggtgagtag
ggaggaagaggtaaaagtttatgcccaaactcttcacacaagagttctagcttacccatt
In the feature-based fasta files, the functional assignment is included as a comment, as shown below.
p3-gto-fasta --protein 1313.7001.gto
>fig|1313.7001.peg.1182 beta-glycosyl hydrolase
MKHEKQQRFSIRKYAVGAASVLIGFAFQAQTVAADGVTTTTENQPTIHTVSDSPQSSENR
TEETPKAELQPETPATDKVASLPKTEEKPQEEVSSTPSDKAEVVTPTSAEKETANKKAEE
ASPKKEEAKEVDSKESNTDKTDKDKPAKKDEAKAEADKPETEAGKERAATVNEKLAKKKI
VSIDAGRKYFSPEQLKEIIDKAKHYGYTDLHLLVGNDGLRFMLDDMSITANGKTYASDDV
KRAIEKGTNDYYNDPNGNHLTESQMTDLINYAKDKGIGLIPTVNSPGHMDAILNAMKELG
IQNPNFSYFGKESARTVNLDNEQAVAFTKALIDKYAAYFAKKTEIFNIGLDEYANDATDA
KGWSVLQADKYYPNEGYPVKGYEKFIAYANDLARIVKSHGLKPMAFNDGIYYNSDTSFGS
FDKDIIVSMWTGGWGGYDVASSKLLAEKGHQILNTNDAWYYVLGRNADGQGWYNLDQGLN
GIKNTPITSVPKTEGADIPIIGGMVAAWADTPSARYSPSRLFKLMRHFANANAEYFAADY
ESAEQALNEVPKDLNRYTAESVAAVKEAEKAIRSLDSNLSRAQQDTIDQAIAKLQETVNN
LTLTPEAQKEEEAKREVEKLAKNKVISIDAGRKYFTLNQLKRIVDKASELGYSDVHLLLG
NDGLRFLLDDMTITANGKTYASDDVKKAIIEGTKAYYDDPNGTTLTQAEVTELIEYAKSK
DIGLIPAINSPGHMDAMLVAMEKLGIKNPQAHFDKVSKTTMDLKNEEAMNFVKALIGKYM
Only protein-encoding genes are output with the --protein option;
however, you see all the features when you use the --feature option.
p3-gto-fasta --feature 1313.7001.gto
>fig|1313.7001.repeat.1 repeat region
tgttttctcaataggattgtactcaggtgagtagggaggaagaggtaaaagtttatgccc
aaactcttcacacaagagttctagcttacccattctatggaatcttgcattatccataat
aataaccgatggtgtggttaatgttggtaagagaaatttctgaaaccatacttcaaaaaa
gtcgctcgtcatcgtctcttcgtaagtcattggagcgattaattcaccatttgttagacc
tgcaaccaaagaaatcctctgatatcttcttccagatactttgcctcttcttaactgacc
ttttaatgagcgaccatattctcgataaaaataagtatcgaatcctgtttcgtcaatcta
aacaggtgctaggtgctttaaactattaaaattcttaagaaataaggctactt
>fig|1313.7001.repeat.2 repeat region
tgttttctcaataggattgtactcaggtgagtagggaggaagaggtaaaagtttatgccc
aaactcttcacacaagagttctagcttacccattctatggaatcttgcattatccataat
Using RAST to Create New Genomes¶
If you have a DNA fasta file and you know the taxonomic ID with a certain degree of confidence, you can use the script p3-rast to annotate the DNA and produce a new genome. The standard output of the script is a GTO. In almost every case, you will want to redirect this to a file. In addition, the new genome is stored in your workspace. It will appear in listings from p3-all-genomes, and you can find its files via the web interface in your QuickData folders.
To invoke p3-rast, you specify a taxonomic ID or the ID of a genome with the same taxonomic ID plus the name to give to the new genome. The contigs should be in the form of a FASTA file via the standard input. All this data is submitted to the BV-BRC annotation service. When the service completes, it stores the new genome in your workspace and sends back a GTO. The example below shows a submission of sequences taken from a metagenomic sample named SRS576036 chosen because they have a high similarity to sequences from Catenibacterium mitsuokai (taxon ID 100886).
p3-rast 100886 "Catenibacterium from sample SRS576036" <sample.fna >test.gto 2>test.log
Now test.gto contains a GTO of the resulting genome and test.log
contains information about the RAST job. If we use the --private
option of p3-all-genomes, we will see the new genome in the list.
p3-all-genomes --private --attr genome_name
genome.genome_id genome.genome_name
100886.26 Catenibacterium from sample SRS576036
The genome was assigned the ID 100886.26. We can see this in the GTO file as well.
{
"genetic_code" : "11",
],
"family_assignments" : [],
"type" : "CDS",
"id" : "fig|100886.26.peg.1540"
},
{
"protein_translation" : "MLQIENASIAYGNDILFSGFNLQLERGEIASISGPSGCGKSSLLNAILGFTPLKEGRIVLNGILLDKGNVDVVRKQTAWIPQELALPLEWVKDMVQLPFGLKANRGTPFSETRLFACFEDLGLEQELYYKRVNEISGGQRQRMMIAVASMIGKPLTIVDEPTSALDSGSAEKVLSFFRRQTENGSAILTVSHDKRFANGCDRHIIMK",
"aliases" : [],
"location" : [
[
"100886.26.con.0010",
"23684",
"-",
624
]
"type" : "CDS"
}
],
"id" : "100886.26",
"contigs" : [
{
"id" : "100886.26.con.0001",
}
The genome ID appears as a part of every feature ID, as an ID in its own right, and as the first part of every contig ID.
As long as you are signed in, the genomes you create using p3-rast will participate in all queries.
p3-all-genomes --eq taxon_id,100886 --attr genome_name
genome.genome_id genome.genome_name
100886.3 Catenibacterium mitsuokai
100886.26 Catenibacterium from sample SRS576036
However, just as you can restrict p3-all-genomes to your own private
genomes using the --private option, you can restrict it to public
genomes only using the --public option.
p3-all-genomes --public --eq taxon_id,100886 --attr genome_name
genome.genome_id genome.genome_name
100886.3 Catenibacterium mitsuokai
The GTO produced by p3-rast has extra information in it describing the annotation process, but it is functionally equivalent to the output were you to re-fetch the genome using the standard script.
p3-gto 100886.26
A p3-gto-scan for test.gto would return the same role profile as for 100886.26.gto.
Customizing Your Toolkit¶
The set of commands that we support via the p3-scripts offers a fairly broad set of capabilities. For example, say you want the name of a specific genome from the ID. You can do this easily using
p3-echo -t genome_id 670.470 | p3-get-genome-data --attr genome_name
genome_id genome.genome_name
670.470 Vibrio parahaemolyticus strain S176-10
If you do this a lot, you may find the extra typing tedious. It is worth, therefore, a brief discussion of how to create shortcut scripts.
Custom Scripts in the BASH Environment¶
In BASH (the most popular Unix shell) you can add functions to your
.bashrc file, using $-notation to indicate the incoming
command-line variables. So, to create the command
gn 670.470
You would use the function definition
function gn {
p3-all-genomes --eq=genome_id,$1 --attr genome_name
}
You must reload the shell to activate your changes to the .bashrc file. Use
exec bash
to replace your current shell with a new instance.
In the function, the $1 is replaced by the first parameter on the
command, which in our example is 670.470. If you type
gn 1313.7001
the $1 is replaced by 1313.7001, so the output would be
genome_id genome.genome_name
1313.7001 Streptococcus pneumoniae P210774-233
You can have more than one parameter. The second is called $2, the
third $3, and so on. The following function creates a genome group
of everything resistant to a particular drug. The drug is the first
parameter, the group name the second.
function rg {
p3-echo -t antibiotic $1 | p3-get-drug-genomes --resistant --attr genome_id | p3-put-genome-group $2
}
Once the above definition is in place, the following command will put all the methicillin-resistant genomes into the group meth_resist.
rg methicillin meth_resist
Custom Scripts for the Windows CMD Shell¶
In Windows, you create a file with the extension .cmd that has your
script in it, and put the file somewhere in your path. The incoming
command-line variables use %-notation. The special command
@echo off is normally put at the beginning of the file to prevent
the file internals from displaying.
So, to create the command
gn 670.470
You would create the file
@echo off
p3-all-genomes --eq=genome_id,%1 --attr genome_name
and save it as gn.cmd in your script directory (which should be some directory you have defined and placed on your path).
In the function, the %1 is replaced by the first parameter on the
command, which in our example is 670.470. If you type
gn 1313.7001
the %1 is replaced by 1313.7001, so the output would be
genome_id genome.genome_name
1313.7001 Streptococcus pneumoniae P210774-233
You can have more than one parameter. The second is called %2, the
third %3, and so on. The following function creates a genome group
of everything resistant to a particular drug. The drug is the first
parameter, the group name the second.
@echo off
p3-echo -t antibiotic %1 | p3-get-drug-genomes --resistant --attr genome_id | p3-put-genome-group %2
Once the above is saved as rg.cmd, the following command will put all the methicillin-resistant genomes into the roup meth_resist.
rg methicillin meth_resist
More Applications¶
The following documents describe more applications for the BV-BRC CLI.
Looking for Hypothetical Proteins in Clusters of Related Features
Computing Signature Clusters: an Application of the Command-Line Tools
What Distinguishes One Set of Genomes from Another? (coming soon)
Uploading Genomes and Assembling Reads (coming soon)